






0 引 言
环氧树脂[1](ER)是综合性能优良、应用广泛 的热固性树脂,但也存在一些缺点,如固化物中存 在大量反应生成的羟基等极性基团,吸水率高、耐 湿性差、电性能不佳和固化树脂脆性大等。为使 ER树脂获得高性能以满足更高的应用要求,人们 不断对ER进行改性,其中一条重要途径就是通过 双马来酰亚胺(BMI)改性ER。
双马来酰亚胺(BMI)因其具有酰亚胺环结构而 具有优异的热稳定性和力学性能[2],且与环氧树 脂有良好的相容性。因而近年来采用BMI改性 环氧树脂研究引起了国内外学者的广泛关注。刘柏 松[4]等人发现,用BMI单体改性环氧树脂、芳香 胺固化体系可以显著提高环氧树脂的韧性和耐热 性。Dae SuKim[5]等人研究了BMI改性环氧树脂体 系的固化行为,并对体系可能发生的化学反应进行 了探讨。
二氮杂萘酮类双酚单体(DHPZ)(见式1)是本 课题组近年来开发的1种新型类双酚单体,具有不 对称扭曲非共平面的结构,以及良好的耐热性和可 溶性。PPEK是由DHPZ经与其他单体聚合得到的 一种新型高分子材料,本文通过在BMI中引入
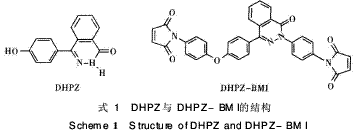
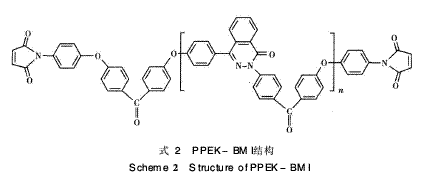
PPEK链段进行扩链,可进一步增大两马来酰亚胺 (M I)间R链的间距,从而适当增大链的自旋和柔软 性,以达到降低固化物的交联密度、减少链的刚性 及改善韧性的目的。因此本文采用自制的含二氮杂 萘酮联苯结构双马来酰亚胺(DHPZ-BMI),即具有 耐高温易溶解的双马来酰亚胺树脂,与DDS形成组 合固化剂对E-51环氧树脂进行固化改性,并将此 三元固化体系与链延长型含醚键双马来酰亚胺树脂 PPEK-BMI(DP=15) /DDS/E-51固化体系作对 比,探讨该体系的固化机理和热稳定性能(见式2)。
1 实验部分
1·1 原 料
E-51环氧树脂:双酚A型,环氧值0·49~0·51; DHPZ-BMI:实验室自制[6]; PPEK-BMI(DP=15): 实验室自制; 4, 4′-二胺基二苯砜(DDS):化学纯, 上海三爱思试剂有限公司生产。
1·2 DHPZ-BMI/DDS/E-51的制备
DHPZ-BMI/DDS以3∶1物质的量比混合均匀 后,再与E-51以1∶1质量比充分混合,置于 130℃油浴中搅拌30 min,得到酒红色的粘稠状产 物,室温下自然冷却。
1·3 固 化
将上述DHPZ-BMI/DDS/E-51置于真空烘箱 除气,按150℃/1 h, 180℃/2 h, 200℃/4 h, 230℃/2 h的固化工艺进行固化,自然冷却至室温。
1·4 分析与表征
使用Nicolet-20DXB型傅里叶变换红外光谱仪, 对固化过程进行跟踪测试;使用NETZSCH DSC204示 差扫描量热仪对树脂固化过程的热效应进行测定, N2气氛,升温速度分别为5, 10, 15℃/min;使 用NETZSCH TGA209型热重分析仪对固化树脂的 热稳定性进行分析, N2气氛,升温速度分别为5, 10, 15℃/min。
2 结果与讨论
2·1 固化过程中的热效应
图1和图2分别为DHPZ-BMI/DDS/E PPEK-BMI/DDS/E-51体系固化进程的曲线 可以看出:固化反应的反应放热峰的位 温速率密切相关,随着升温速率的提高,体 起始温度和峰值温度均提高,固化时间缩短 位时间产生的热效应增大,固化反应放热峰 向高温移动。
对图1采用Kissinger[7]方程计算固化反 化能与频率因子。假设固化反应最大速率发 化反应放热峰的峰顶温度,反应级数在固化
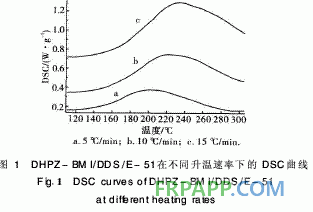

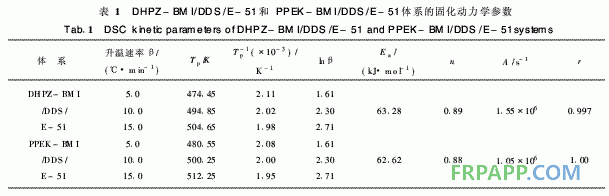
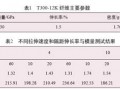
DHPZ-BMI/DDS/E-51体系的DSC数据及 固化反应动力学参数汇总见表1。同样采用Kissi ger方程和Crane方程对图2计算其固化反应表观 活化能、表观频率因子、线性相关系数和反应级 数。由表1可见DHPZ-BMI/DDS/E-51和PPE -BMI/DDS/E-51两种体系的固化反应活化能几 乎相同,说明二者的固化反应过程相同,且固化反 应活化能均较低、频率因子较大,故二者固化反应 速率均较快,与两马来酰亚胺(MI)间R链的间距 无关。
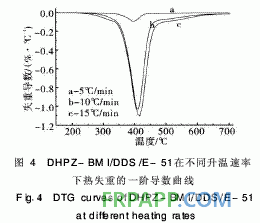
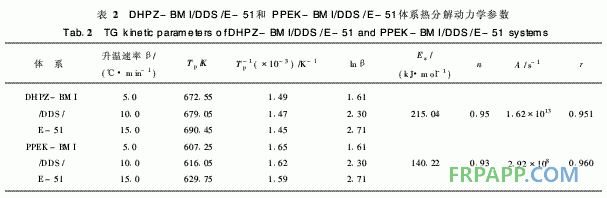
~440℃之间,起始分解温度为260℃, 5%热失 重温度为341℃,该阶段的热分解失重可能是分子 间的弱键,如C—O—C的分解;第2阶段在440℃ 之后,这一阶段失重则是由于苯环、酰亚胺等裂解 造成的,且失重率明显减小,再次证明固化体系良 好的热稳定性。从图4取DTG曲线第1个峰的峰 顶温度进行比较。按2·1节中的方法求取的热分解 动力学参数见表2。
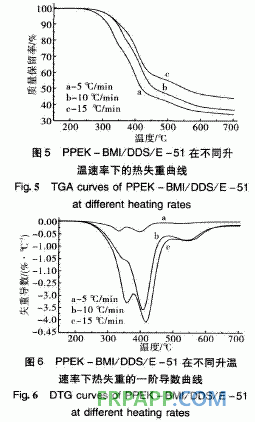
由图5可知,其热失重也分为2个阶段:第 1阶段在240~420℃之间,起始温度为240℃, 5%热失重温度为298℃;第2阶段在420℃之 后,失重率也明显减小。从图6取固化树脂的 DTG第一峰顶温度依Kissinger和Crane方程进行 计算,得到的固化树脂的热分解动力学分解参数 如表2所示。
表2中2种固化树脂的热分解活化能存在明显 差异, DHPZ-BMI/DDS/E-51固化树脂的分解活 化能较高,为PPEK-BMI/DDS/E-51固化树脂的 1·5倍以上,这是因为DHPZ-BMI/DDS/E-51固 化树脂中二氮杂萘联苯结构比例高,耐热性好,而 PPEK-BMI/DDS/E-51固化树脂中有大量醚键存 在,降低了二氮杂萘联苯结构在固化树脂中的比 例,导致耐热性下降,因此其起始分解温度低,相 同热失重的分解温度也较低。
3 结 论
1)由DHPZ-BMI/DDS/E-51和PPEK-BMI/ DDS/E-51体系的固化反应动力学数据相比得知, 这2种体系的表观活化能几乎相同,说明二者的固 化反应过程相同,且固化反应活化能均较低、频率 因子较大,故二固化反应速率均较快,与两马来酰 亚胺(MI)间R链的间距无关。
2)DHPZ-BMI/DDS/E-51和PPEK-BMI/ DDS/E-512种固化树脂的热分解活化能存在显著 差别, DHPZ-BMI/DDS/E-51固化树脂的热分解 活化能达215·04 kJ/mo,l是PPEK-BMI/DDS/E- 51固化树脂的1·5倍以上,这是因为DHPZ- BMI/DDS/E-51固化树脂中二氮杂萘联苯结构比 例高,耐热性好,而PPEK-BMI/DDS/E-51固化 树脂中有大量醚键存在,降低了二氮杂萘联苯结构 在固化树脂中的比例,导致耐热性下降。亦即表 明:采用DHPZ-BMI/DDS组合固化剂固化的E- 51型环氧树脂热稳定性更佳。







 鲁ICP备2021047099号
鲁ICP备2021047099号